|
|
|



The efficacy and safety of 24-hour continuous levodopa-based therapy with PRODUODOPA were demonstrated in 2 trials.1-3

12-Week
Pivotal Phase 3, randomized, double-blind, double-dummy, active-controlled study evaluating the efficacy and safety of 24‑hour daily continuous subcutaneous infusion of PRODUODOPA compared with oral IR levodopa/carbidopa1,3

52-Week
Phase 3, single-arm, open-label study evaluating the safety and efficacy of 24-hour daily exposure with a continuous subcutaneous infusion of PRODUODOPA1,2
IR=immediate-release.
Aim: Evaluate the efficacy, safety, and tolerability of PRODUODOPA to oral IR levodopa/carbidopa in patients with advanced PD.
Duration: 12 weeks.
Study type: Phase 3, 1:1 randomized, double-blind, double-dummy, active-controlled.
Patient population: Evaluation of 141 patients. The target population was levodopa-responsive patients with advanced PD whose motor symptoms were inadequately controlled with current treatment and experienced minimum daily average of 2.5 hours of “Off” time per day, as assessed by PD diaries.*
Of the 74 patients randomized to receive PRODUODOPA, 48 completed the study and 26 discontinued treatment.
Of the 67 patients receiving optimized oral IR levodopa/carbidopa, 62 completed treatment and 5 discontinued.
*Based on the PD diary, the average daily normalized “On” time for patients on PRODUODOPA was 9.20 (+/-2.42) hours at baseline and increased by 2.72 (+/-0.52) hours at Week 12 compared with an increase of 0.97 (+/-0.50) hours at Week 12 from a baseline of 9.49 (+/-2.62) hours for patients taking optimized IR levodopa/carbidopa (LS mean change [SE]). This resulted in a statistically significant improvement of 1.75 (+/-0.65) hours in patients on PRODUODOPA vs oral IR LD/CD (LS mean of difference [SE]; P=0.0083). Average daily normalized “Off” time for patients on PRODUODOPA was 6.34 (+/-2.27) hours at baseline and decreased by 2.75 (+/-0.50) hours at Week 12 compared with a decrease of 0.96 (+/-0.49) hours at Week 12 from a baseline of 5.91 (+/-1.88) hours for patients taking optimized IR levodopa/carbidopa (LS mean change [SE]). This resulted in a statistically significant improvement of 1.79 (+/-0.63) hours in patients on PRODUODOPA vs oral IR LD/CD (LS mean of difference [SE]; P=0.0054).1
IR=immediate-release; LD/CD=levodopa/carbidopa; PD=Parkinson’s disease; LS=least squares; SE=standard error.
What Does 24-Hour Continuous Delivery Without Surgery Mean for Your Patients?


Reduction in morning "Off" state (morning akinesia)1*

Fewer interruptions over 24 hours morning, day, and night3*

More “On” time and less “Off” time over 24 hours means patients can do more1†

*Patients recorded their PD symptoms in a PD Diary, which was completed for a full 24-hour period. Patients made an entry upon waking and every 30 minutes during their normal waking time and upon awakening from time asleep. Patients recorded their "On" time without troublesome dyskinesia (sum of "On" time without dyskinesia and "On" time without troublesome dyskinesia) and "Off" time as assessed by the PD Diary.3
†More refers to improvement for patients on PRODUODOPA compared with oral IR levodopa/carbidopa in “On” and “Off” time at Week 12.3
IR=immediate-release; PD=Parkinson's disease.
Change from baseline to Week 12 in “On” time without troublesome dyskinesia*




(LS mean of difference=1.75 hours; P=0.0083)
*Based on the PD diary, the average daily normalized “On” time without troublesome dyskinesia for patients on PRODUODOPA was 9.20 (SD +/-2.42) hours at baseline and increased by 2.72 (SE +/-0.52) hours at Week 12 compared with an increase of 0.97 (SE +/-0.50) hours at Week 12 from a baseline of 9.49 (SD +/-2.62) hours for patients taking optimized IR levodopa/carbidopa (LS mean change [SE]). This resulted in a statistically significant improvement of 1.75 (SE +/-0.65) hours in patients on PRODUODOPA vs oral IR LD/CD (LS mean of difference [SE]; P=0.0083). “On” time without troublesome dyskinesia is the sum of “On” time without dyskinesia and “On” time with non‑troublesome dyskinesia.
†Model-based least square mean (standard error) of change.
‡Calculated by dividing the change in hours from baseline by the number of hours reported at baseline.
IR=immediate-release; LS=least squares; PD=Parkinson's disease; SD=standard deviation; SE=standard error; LD/CD=levodopa/carbidopa.
Change from baseline to Week 12 in “Off” time*




(LS mean of difference=-1.79 hours; P=0.0054)
*Based on the PD diary, the average daily normalized “Off” time for patients on PRODUODOPA was 6.34 (SD +/-2.27) hours at baseline and decreased by 2.75 (SE +/-0.50) hours at Week 12 compared with a decrease of 0.96 (SE +/-0.49) hours at Week 12 from a baseline of 5.91 (SD +/-1.88) hours for patients taking optimized IR levodopa/carbidopa (LS mean change [SE]). This resulted in a statistically significant improvement of 1.79 (SE +/-0.63) hours in patients on PRODUODOPA vs oral IR LD/CD (LS mean of difference [SE]; P=0.0054).
†Model-based least square mean (standard error) of change.
‡Calculated by dividing the change in hours from baseline by the number of hours reported at baseline.
IR=immediate-release; LD/CD=levodopa/carbidopa; LS=least squares; PD=Parkinson's disease; SD=standard deviation; SE=standard error.
Note to affiliate: Morning akinesia has been included within the Study 736 publication and is being reviewed as part of the "Type II variation SmPC assessment" by MPA. Please assess inclusion of morning akinesia information data based on your local rules and regulations
Percentage of patients waking up in the “On” state*†‡




Secondary efficacy endpoints were tested in hierarchical order. Hierarchical testing ended after the first secondary endpoint as the next prespecified endpoint did not reach significance.3 PRODUODOPA demonstrated improvements at Week 12 in morning akinesia compared to baseline, but did not achieve statistical significance when compared to active control arm.1
*Morning akinesia was defined as reporting “Off” status as the first morning symptom upon awakening.
†Percentage of patients without morning akinesia is the sum of patients reporting “On” time without dyskinesia, “On” time with non-troublesome dyskinesia, and “On” time with troublesome dyskinesia upon awakening.
‡Based on the PD diary, patients on PRODUODOPA, “On” without dyskinesia was reported in 70% (33/47) of patients; “On” with nontroublesome dyskinesia was reported in 8% (4/47) of patients; “On” with troublesome dyskinesia was reported in 4% (2/47) of patients. For patients on oral IR LD/CD “On” without dyskinesia was reported in 37% (22/60) of patients. Based on the PD diary, 79% (56/71) of patients on PRODUODOPA experienced morning akinesia at baseline and decreased to 17% (8/47) of patients at Week 12. By comparison, 76% (51/67) of patients taking optimized oral IR LD/CD experienced morning akinesia at baseline and decreased to 63% (38/60) of patients at Week 12.
§Calculated by dividing the change in hours from baseline by the number of hours reported at baseline.
IR=immediate-release; LD/CD=levodopa/carbidopa; PD=Parkinson’s disease.
Aim: Evaluate the safety and tolerability of 24-hour daily exposure with a continuous subcutaneous infusion of PRODUODOPA. Secondary efficacy endpoints were also evaluated.
Duration: 52 weeks (this study is ongoing).
Study type: Phase 3, single-arm, open-label.
Patient population: Evaluation of 244 patients. 137 patients completed the 52-week study. The target population was levodopa-responsive patients with PD whose motor symptoms were inadequately controlled with current treatment and experienced a minimum of 2.5 hours of “Off” time per day as assessed by PD diaries.*
PD=Parkinson’s disease.




The PRODUODOPA safety study is ongoing. Data presented reflect the third interim analysis of 52-week study results that include 104 patients. “On” time without troublesome dyskinesia improved by an average of 3.8 (+/-3.3) hours by Week 52 (average 12.9 hours*) compared to baseline (average 9.1 [+/-2.5] hours*; n=236)
(P≤0.001) based on the PD diary.1,2
*Values represent unadjusted mean hours (+/-standard deviation) assessed using a 24-hour PD diary and normalised to a 16-hour waking day. Only patients who completed each study visit were included in the efficacy analysis, and no adjustments were made to account for premature discontinuation.
†Calculated by dividing the change in hours from baseline by the number of hours reported at baseline.




The PRODUODOPA safety study is ongoing. Data presented reflect the third interim analysis of 52-week study results that include 104 patients. “Off” time was decreased by an average of 3.5 (+/-3.1) hours by Week 52 (average 2.4 hours) compared to baseline (average 5.9 [+/-2.2] hours*; n=236) (P≤0.001) based on the PD diary.1,2
*Values represent unadjusted mean hours (+/-standard deviation) assessed using a 24-hour PD diary and normalised to a 16-hour waking day. Only patients who completed each study visit were included in the efficacy analysis, and no adjustments were made to account for premature discontinuation.
†Calculated by dividing the change in hours from baseline by the number of hours reported at baseline.
Continue to Safety page
Please refer to the PRODUODOPA SmPC for complete Prescribing and Safety Infomation.

Levodopa exposure following 24-hour PRODUODOPA infusion and 16-hour DUODOPA infusion followed by nighttime oral LD/CD doses5
Formulation

PRODUODOPA

DUODOPA + Nighttime Oral LD/CD
Note to affiliate: the graph presented here is based on the published Rosebraugh reference, however the colors have been adjusted to match the project. Please update per your local rules and regulations.
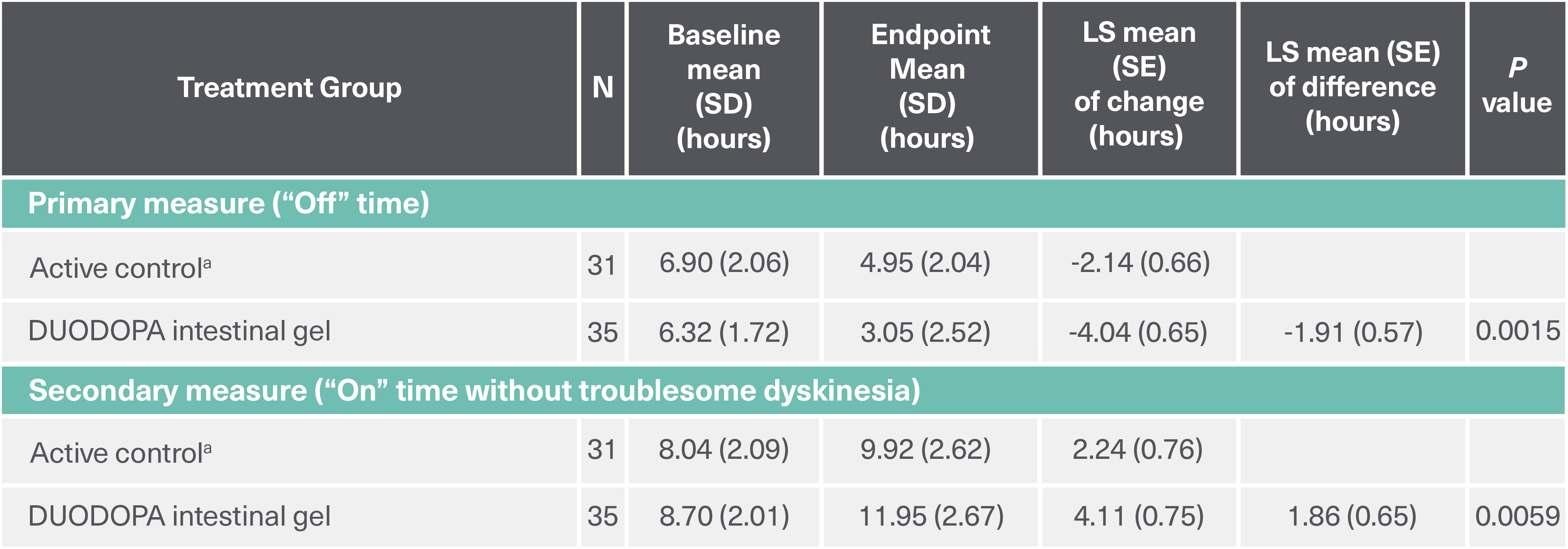
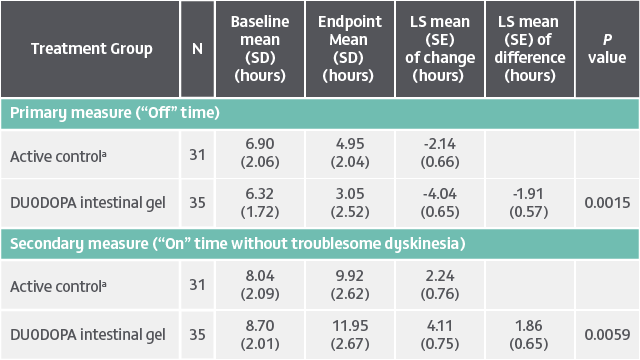
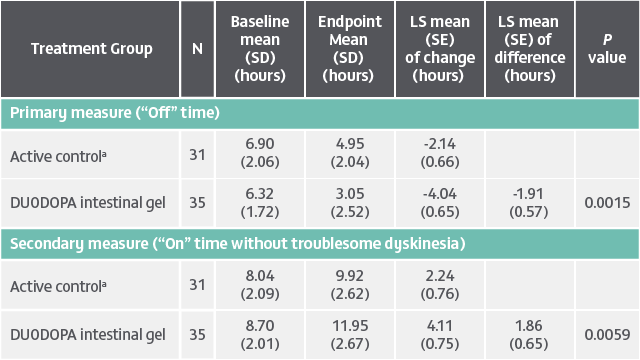
DUODOPA demonstrated significant improvements in “Off” time (baseline to endpoint) and “On” time without troublesome dyskinesia (baseline to endpoint) compared to oral levodopa/carbidopa (LS mean difference). Analysis of other secondary efficacy endpoints are not presented here. Please refer to the PRODUODOPA SmPC for further information about these endpoints.
Duration: 12 weeks. Study type: Randomized, double-blind, double-dummy, double-titration trial. Patient population: 71 adult patients with advanced PD and motor complications. Aim: To assess the efficacy and safety of levodopa-carbidopa intestinal gel delivered continuously through an intrajejunal percutaneous tube. Primary endpoint: Change from baseline to final visit in motor “Off” time.4
<placeholder for DUODOPA Indication and Summary of Important Treatment Considerations>
<placeholder link for local DUODOPA SmPC>
LS=least squares; SD=standard deviation; SE=standard error.
aActive control, oral levodopa/carbidopa 100/25 mg tablets.